
Packaging a uso farmaceutico: analisi TOC secondo USP 661
Il packaging è un elemento comune a svariati settori industriali, spaziando dai noti MOCA nel campo alimentare fino a settori più specifici come l’edilizia, la cosmetica e, naturalmente, il farmaceutico.
La sua importanza è cruciale per chi produce il contenuto, dovendo garantire la conservazione delle caratteristiche del prodotto. Nel caso degli alimenti, oltre a mantenerli in condizioni stabili, il packaging deve prevenire la contaminazione del prodotto, prima di qualunque altra cosa.
In settori come quello cosmetico, il packaging non solo deve svolgere una funzione pratica ma deve anche attrarre il consumatore, richiedendo una progettazione attenta sia agli aspetti estetici che a quelli pratici.
Nel settore farmaceutico, il packaging si distingue per le sue prerogative uniche, regolamentate da rigide normative che disciplinano ogni aspetto, dai materiali utilizzati per gli imballaggi all’etichettatura. La prima e fondamentale considerazione riguarda l’imballaggio stesso, che deve garantire il rispetto di specifiche caratteristiche.
Il packaging nel settore farmaceutico
La perfetta adattabilità al medicinale è un requisito essenziale, poiché l’imballaggio deve salvaguardare l’integrità del prodotto e prevenire l’ingresso di agenti esterni nocivi. La resistenza alle condizioni ambientali esterne è altrettanto cruciale, concentrandosi sulla necessità di un packaging primario che sia resistente alla luce e all’aria, per preservare la stabilità del farmaco.
Le capacità protettive costituiscono un ulteriore elemento chiave: l’imballaggio primario deve rimanere incolume durante i vari spostamenti del prodotto lungo la catena di distribuzione, garantendo che il farmaco arrivi al consumatore in condizioni ottimali.
La comunicazione trasparente è un aspetto altrettanto importante. Un packaging a norma deve fornire indicazioni chiare e dettagliate sulla natura del contenuto e sulle modalità di somministrazione del medicinale, consentendo al paziente di utilizzare il prodotto in modo corretto e sicuro.
Normative per il packaging farmaceutico: USP 1663 e 661
Affinché un materiale plastico sia considerato conforme per l’utilizzo farmaceutico vengono eseguiti numerosi test sul materiale sia per le sostanze estraibili sia per le sostanze rilasciabili. Le sostanze estraibili sono i composti che possono essere estratti dal contenitore, mentre quelli rilasciabili possono penetrare nella formulazione quando sono in contatto diretto con il farmaco.
Una delle più recenti linee guida riguardanti gli estraibili è la USP <1663>, che fornisce dettagli su come il packaging farmaceutico e i sistemi di somministrazione dei farmaci possano essere valutati attraverso test E&L (Extractables & Leachables).
Oltre alle indicazioni fornite dalla USP <1663>, i produttori possono seguire ulteriori linee guida al fine di garantire l’aderenza alle migliori pratiche nello sviluppo di prodotti farmaceutici e nell’imballaggio di dispositivi medici.
Di particolare rilievo per le normative sugli imballaggi farmaceutici è l’USP <661>, che stabilisce i test correlati e i criteri di accettazione per gli imballaggi contenenti prodotti farmaceutici, biologici, integratori alimentari e dispositivi.
Per creare la specificità richiesta per la qualità e la sicurezza dell’imballaggio farmaceutico, l’USP ha ritenuto necessario creare due sezioni del 661:
- La norma USP 661.1 riguarda i materiali plastici da costruzione
- La norma USP 661.2 si concentra sull’idoneità/sicurezza del sistema di imballaggio completo
Nel periodo precedente al 1° dicembre 2025, le aziende farmaceutiche che rilasciano i propri prodotti possono scegliere di conformarsi alle linee guida stabilite nell’attuale USP 661 oppure scegliere di conformarsi sia a USP 661.1 che a USP 661.2.
Cosa succede dopo il 1 dicembre 2025
Dopo il 1° dicembre 2025, l’unica opzione disponibile sarà la conformità alla norma USP 661.2, idealmente integrata dalla conformità alla norma USP 661.1. Il rispetto di entrambi non è necessario, ma potrebbe rivelarsi utile poiché i revisori della FDA potrebbero richiedere una documentazione più ampia.
All’interno di queste due sezioni sono presenti le migliori pratiche per eseguire le analisi TOC sugli estratti dei materiali plastici utilizzati per i componenti con i quali vengono prodotti i packaging farmaceutici. Le sezioni volgono a migliorare la sicurezza dei prodotti farmaceutici, promuovendo l’utilizzo di materiali caratterizzati in modo completo ed efficiente. La determinazione del TOC è solo uno dei parametri considerati, insieme alla biocompatibilità, alle proprietà fisico-chimiche, agli additivi plastici e ai metalli estraibili.
Secondo le normative USP 661.1 e 661.2, la determinazione del TOC richiede un intervallo dinamico lineare di 0,2–20 mg/l, con un limite di rilevabilità di 0,2 mg/l. Il valore limite di TOC nei componenti per il packaging è NMT (Not More Than) 5 mg/l (USP 661.1), mentre nel packaging in plastica è NMT 8 mg/l (USP 661.2).
Fase analitica – TOC
L’estrazione dei materiali di imballaggio è descritta nelle linee guida della USP ed è specifica per tipologia di materiale da testare. Una volta preparato il campione deve essere analizzato entro 4 ore, come descritto nel capitolo 643 della USP. Questo capitolo è completamente dedicato all’analisi del TOC, sia per qualificare la tecnica analitica sia per fornire indicazioni sull’interpretazione dei dati ai limiti del range di quantificazione.
Tra gli strumenti per l’analisi TOC da estratti di materiali plastici ad uso farmaceutico possono essere utilizzati sia sistemi con ossidazione con lampada UV sia con combustione catalitica ad elevata temperatura. La serie multiNC pharma di Analytik Jena offre entrambe le soluzioni con gli analizzatori TOC multiNC pharma UV e multi N/C 3100 Pharma, che si equivalgono da un punto di vista dei risultati analitici.
Analytik Jena ha messo a confronto le due tecniche sui seguenti materiali plastici:
- Polietilene, olefine cicliche e polipropilene;
- Polietilene tereftalato e polietilene tereftalato glicole (PET-G)
- Cloruro di polivinile plasticato.
I campioni sono stati preparati nella seguente modalità:
Polietilene, olefine cicliche e polipropilene
25 g di materiale vengono posti in un pallone di vetro borosilicato e fatti bollire a riflusso con 500 ml di acqua ultrapura per 5 ore. La soluzione estratta raffreddata viene filtrata e raccolta in un matraccio da 500 ml e poi portato a volume con acqua. Questa soluzione deve essere utilizzata entro 4 ore dalla preparazione per la determinazione del TOC.
Polietilene tereftalato e polietilene tereftalato glicole (PET-G)
10 g del materiale vengono posti in un pallone di vetro borosilicato e riscaldati a 50 °C con 200 ml di acqua ultrapura per 5 ore. Dopo raffreddamento, la soluzione viene travasata in un matraccio da 200 ml e portata a volume con acqua. Questa soluzione deve essere utilizzata entro 4 ore dalla preparazione per la misura del TOC.
Cloruro di polivinile plastificato
25 g del materiale vengono posti in un pallone di vetro borosilicato e vengono aggiunti 500 ml di acqua ultrapura e il collo del pallone viene coperto con un foglio di alluminio. Il pallone viene riscaldato in un’autoclave a 121 ± 2 °C per 20 minuti. Dopo raffreddamento, la soluzione viene travasata in un matraccio da 500 ml e portata a volume con acqua. Questa soluzione deve essere utilizzata entro 4 ore dalla preparazione per la misura del TOC.
Packaging in plastica per uso farmaceutico
Il packaging viene riempito alla sua normale capacità con acqua ultrapura e chiuso. Il sistema viene riscaldato in autoclave a 121 ± 2 °C per 30 minuti. Se il riscaldamento a 121 °C porta al deterioramento del contenitore, si può applicare un trattamento termico a 100 ± 2 °C per 2 ore o a 70 ± 2 °C per 24 ± 2 ore. Dopo il raffreddamento, il contenuto viene analizzato entro 4 ore dalla preparazione.
Calibrazione
Entrambi gli analizzatori, sia con ossidazione UV che con combustione catalitica ad elevata temperatura, vengono calibrati per NPOC nell’intervallo da 0,1 a 20 mg/l con soluzioni standard preparate da una soluzione madre di saccarosio da 1000 mg/l.
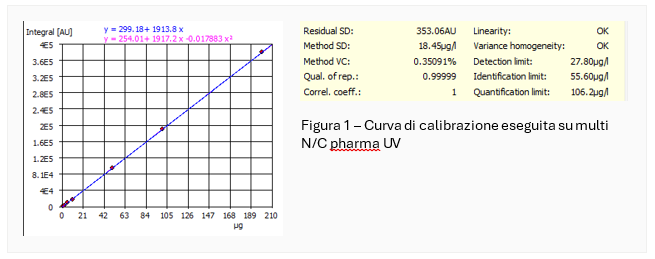
Entrambe le calibrazioni mostrano un’eccezionale linearità nell’intero intervallo di calibrazione. A titolo di esempio, si riporta la calibrazione effettuata con il multi-NC pharma UV.
Strumentazione
L’analisi è stata eseguita con multi NC pharma UV, con multi NC pharma HT e con il sistema multi NC 3100 pharma. I 3 sistemi sono utilizzabili in modo equivalente per questa applicazione. Questi sistemi si differenziano tra loro per alcune caratteristiche che li rendono più o meno adatti ad alcune tipologie di analisi. La prima differenza è nella modalità di ossidazione. Il modello multi NC pharma UV utilizza l’ossidazione con lampada UV, mentre gli altri 2 sistemi sono tutti caratterizzati dall’ossidazione ad elevata temperatura. Un’altra differenza importante è sicuramente il volume massimo iniettabile. Per il sistema multi NC pharma UV abbiamo un volume massimo iniettabile di 20ml, per multi NC pharma HT 3ml e infine per multi NC 3100 pharma 1ml.
Tre estratti da materiali plastici utilizzati per il packaging sono stati analizzati insieme a uno standard di controllo dopo la calibrazione del sistema, come descritto. I risultati sono visualizzati nella Tabella 1.
Campione |
media NPOC (mg/l) |
RSD (%) |
Campione 1 |
2.48 |
0.9 |
Campione 2 |
0.984 |
1.3 |
Campione 3 |
8.72 |
0.5 |
QC (2.0 mg/l) |
2.04 |
0.7 |
Questa nota applicativa dimostra che gli analizzatori TOC della serie multi N/C pharma forniscono le caratteristiche prestazionali richieste per la conformità agli standard USP per le analisi TOC nel packaging farmaceutico e nei suoi componenti.


